LAMMPS模拟软件的功能有很多:
微观结构演化
结构优化
过渡态搜索
热导率计算
等等(太多了,有兴趣的可自行百度)。。。。。
其中它最常用的功能就是模拟微观结构的演化,即观察粒子在合理物理环境(符合牛顿定律)下的演化情况。打个比方,我们高中时期学物理时经常会碰到这样的一个问题:两个人在操场跑圈,一个速度快一个速度慢,那么他们在什么时候会第一次相遇,或者说1个小时内会相遇几次?然后我们就会根据公式去哗啦啦的计算,最后得到结果。而LAMMPS软件的作用就是,我们把题目中的物理条件和环境都告诉它,它来帮我们计算,最后我们便能轻松的能通过它输出的结果(LAMMPS不帮助处理数据,LAMMPS的所有输出都需要自己处理)看到在操场跑圈的两个人的相遇情况。
LAMMPS的输入文件:
File_name.in(俗称in文件,必备)
Data文件(模型文件,可有可无)
势文件或力场文件(可有可无,根据力场类型的选择确定)
LAMMPS输出文件:
默认输出log文件帮助查看运行过程是否正常,其它输出则需要自行在in文件中自行定义,否则无输出~
一般来说,in文件内容的主要框架包含以下几个部分:
初始模拟系统设置
初始模型构建(读取模型数据)
定义原子间相互作用势或设置力场(势文件或力场文件)
定义原子/体系某些信息的计算(原子势能。。)
定义输出原子(坐标)/体系(热力学)信息
模拟环境设定并运行
建议:in文件一定自己写!!!就算有借鉴的in文件(例如百度得来的,师兄师姐留下来的。。),也要自己重写一遍,这样不仅能更好的理解in文件里面的内容,而且一旦后续模拟出问题了,自己也能更快的找到问题所在。
先看一个用LAMMPS做的简单的MD模拟——bcc钨的弛豫过程
(弛豫过程:一般进行复杂模拟前必备的过程,目的是使模型达到热力学平衡状态。)
1.输入文件in.relax内容
初始模拟系统设置
units metal
atom_style atomic
boundary p p p初始模型构建(读取模型数据)
lattice bcc 3.168
region box block 0 5 0 5 0 5
create_box 1 box
create_atoms 1 box定义原子间相互作用势
pair_style eam/alloy
pair_coeff W_zhou.eam.alloy W设定计算原子势能
compute 1 all pe/atom
定义输出原子(坐标)/体系(热力学)信息
thermo 10
thermo_style custom step temp pe etotal dt time
dump 1 all custom 100 W.xyz id type x y z c_1模拟环境设定并运行(NVT系综下弛豫)
velocity all create 300 666 mom yes rot yes dist gaussian
fix 1 all nvt temp 300 1000 1
timestep 0.001
run 10000
2.由于我们直接在in文件中构建好了模型,所以不需要data文件
3.由于我们采用了eam相互作用势(最适合金属材料的相互作用势),所以需要势文件(W_zhou.eam.alloy,这个可以在LAMMPS软件包中的Potential文件下找到,找到后复制这个文件到你的in文件的目录下,不要让它和in文件分开),这个势文件的作用是告诉LAMMPS如何计算体系中原子受力的,后续文章会详细介绍,现在知道即可。
4.在in文件所在目录下输入(并行)命令:mpirun-n 2 lmp_mpi < in.relax 其中2 为调用的核数即可开始计算。放后台运算的命令为:mpirun-n 2 lmp_mpi < in.relax > out.dat &
5.百度与LAMMPS软件绝配的数据后处理软件OVITO,从官网上下载最新版的OVITO软件(往后文章中所涉及到的例子都会利用到OVITO软件),然后先尝试下自己将LAMMPS输出的W.xzy文件导入进去,观看整个弛豫过程。可以看到如下动态过程: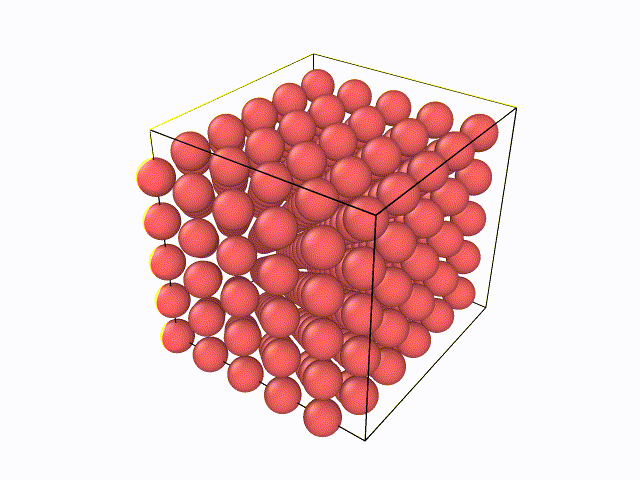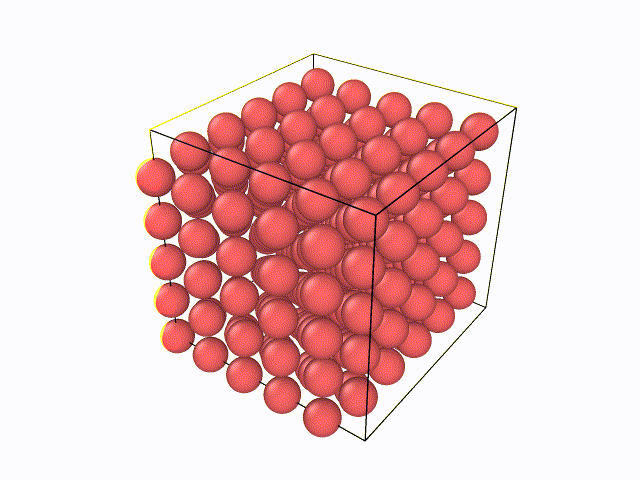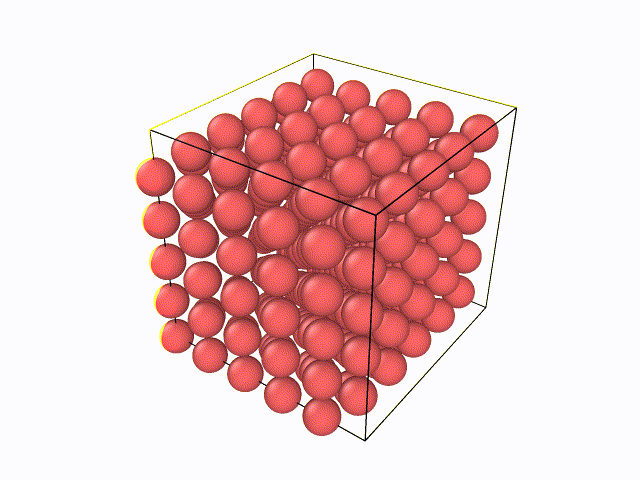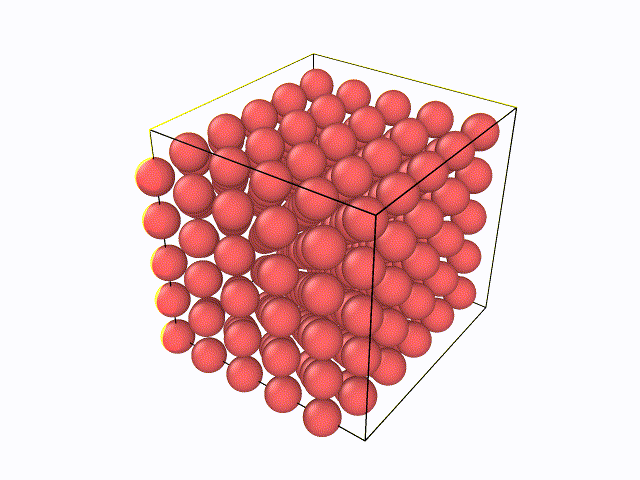
刚开始学习的同学可以先依葫芦画瓢完成这一个简单的模拟,并尝试着理解下in文件中的内容,后面我们会结合LAMMPS权威手册对in文件中的这6个主要内容进行详细讲解,并逐步帮助新手们养成阅读手册的好习惯。另外,建议新手入手一门编程语言(现在Python很火,可以考虑一波,fortran,C之类的也可以,至少会一个,不需要很精通,当然精通会更好~),虽然学习LAMMPS可能用不到,但是处理数据和建模时,会编程会方便很多。
由于本系列的侧重点在于LAMMPS软件的使用,所以关于分子动力学的原理,本系列不会过多深入的介绍(只会在有必要时提及),喜欢深入研究理论的同学考虑研究以下书籍:
英文书籍:Understandingmolecular simulation: From algorithms to applications (这本书最好能找到原版,我之前没办法看的盗版,很多错误,看的心很累)和Molecular Modelling - Principles and Applications。
中文书籍:《分子模拟的理论与实践》(这本书希望你们看的时候不会瞌睡~)
转载请注明来源,欢迎对文章中的引用来源进行考证,欢迎指出任何有错误或不够清晰的表达。可以在下面评论区评论,也可以邮件至 weig16@whu.edu.cn

